

胆囊收缩素通过抑制小胶质细胞对兴奋性突触的吞噬作用改善脓毒症相关脑病小鼠认知功能障碍
期刊: Journal of Neuroinflammation
作者:Lei Chen等
分区:中科院一区
影响因子:10.1
本研究首次明确了胆囊收缩素(CCK)通过抑制小胶质细胞补体1q(C1q)介导的兴奋性突触吞噬,改善脓毒症相关脑病(SAE)小鼠认知障碍的分子机制,同时证实外源性CCK补充和内源性CCK阳性神经元激活均为SAE潜在治疗策略,为SAE的认知障碍干预提供了全新的靶点和实验依据。
一、研究背景与科学问题
1. 研究背景
败血症相关性脑病(SAE)以认知障碍为特征,是败血症患者常见的并发症。小胶质细胞通过吞噬突触参与各种认知障碍相关疾病。胆囊收缩素(CCK)是大脑中丰富的神经肽,与认知功能密切相关。然而,CCK在SAE中的作用以及CCK与突触小胶质吞噬作用之间的关系尚不清楚。
2. 核心科学问题
CCK是否能改善SAE小鼠的认知障碍?
CCK若发挥神经保护作用,是否通过抑制小胶质细胞C1q介导的兴奋性突触吞噬实现?
CCK发挥作用的受体亚型及细胞靶点是什么?
激活内源性CCK阳性神经元是否与外源性CCK补充具有相似的治疗效果?
二、研究方法
脂多糖(LPS)被用于构建3个月龄雄性小鼠和BV2小胶质细胞的SAE模型。为研究CCK对SAE模型小鼠认知障碍的影响,我们采用外源性CCK注射至背海马CA1区域,或通过化学激活CCK阳性神经元,促进内源性CCK释放。使用莫里斯水迷宫和恐惧条件反射测试来评估小鼠的认知功能。进行了RNA测序以探索CCK诱导神经保护的潜在信号通路。采用西方印迹和免疫荧光法评估CCK对突触、神经毒性星形胶质细胞和兴奋性突触小胶质细胞吞噬作用的影响。采用全细胞记录方法确定兴奋性突触传递。
莫里斯水迷宫(MWM):检测海马依赖性空间学习记忆,记录逃避潜伏期、平台穿越次数、目标象限停留时间,排除游泳速度对结果的干扰;
恐惧条件化实验(FCT):分为场景测试(海马依赖性)和声音测试(非海马依赖性),记录小鼠冻结时间占比。
三、研究结果
1. SAE小鼠出现认知障碍,且伴随海马CCK降低和兴奋性突触丢失
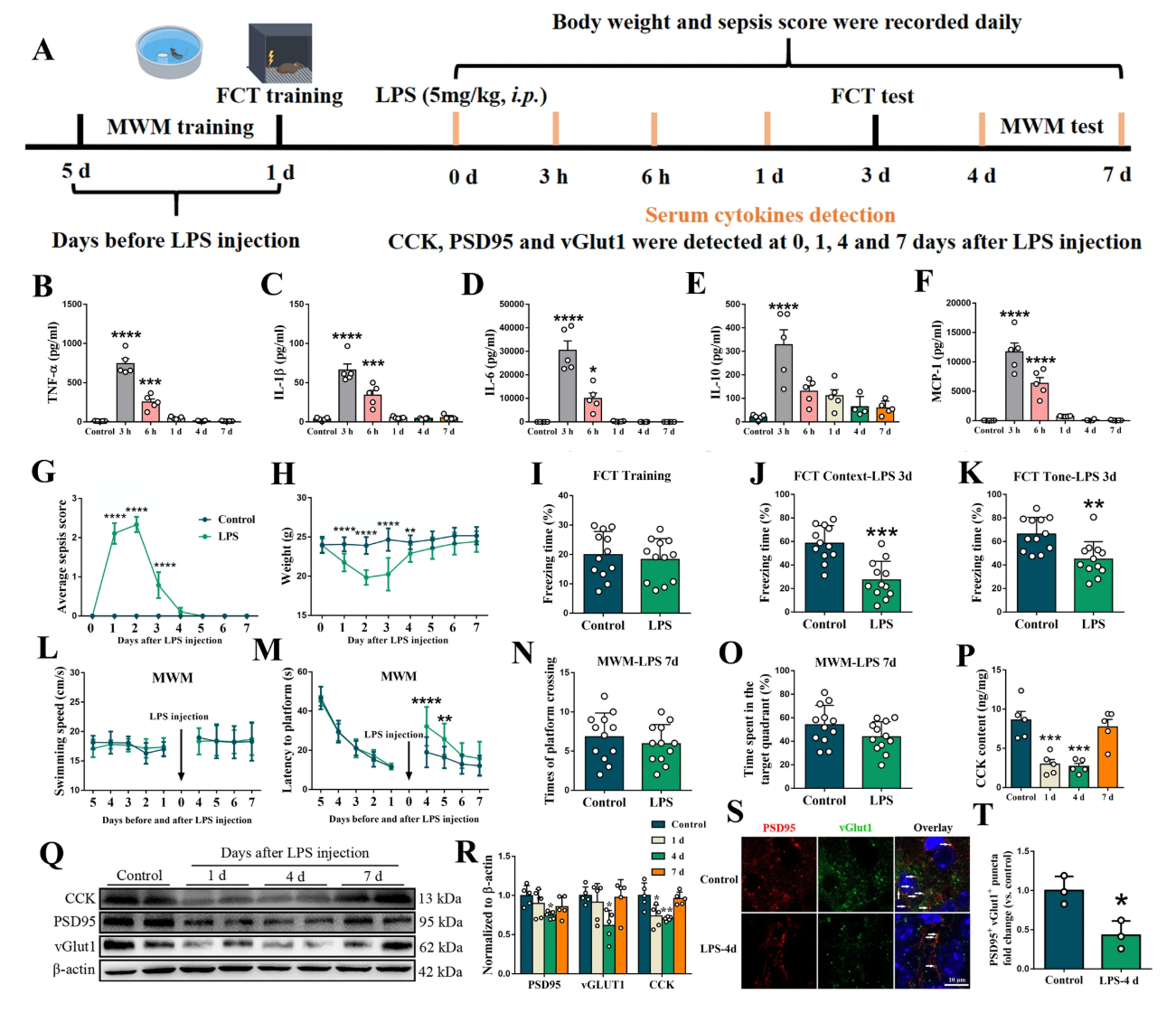
LPS造模后小鼠血清炎症因子(TNF-α、IL-1β、IL-6、IL-10、MCP-1)在3/6h显著升高,脓毒症评分1~3d升高、体重1~4d降低,SAE模型构建成功;
认知功能:FCT显示造模后3d小鼠场景和声音测试冻结时间均降低(海马/非海马依赖性记忆均受损);MWM显示造模后4/5d逃避潜伏期延长,7d认知功能部分恢复,提示SAE小鼠存在短期认知障碍;
分子变化:造模后1/4d海马CCK蛋白水平显著降低,兴奋性突触标志物(PSD95、vGlut1)蛋白水平及突触数量显著减少,提示CCK降低与突触丢失、认知障碍同步发生。
2. 海马CA1区注射CCK8可改善SAE小鼠的认知障碍
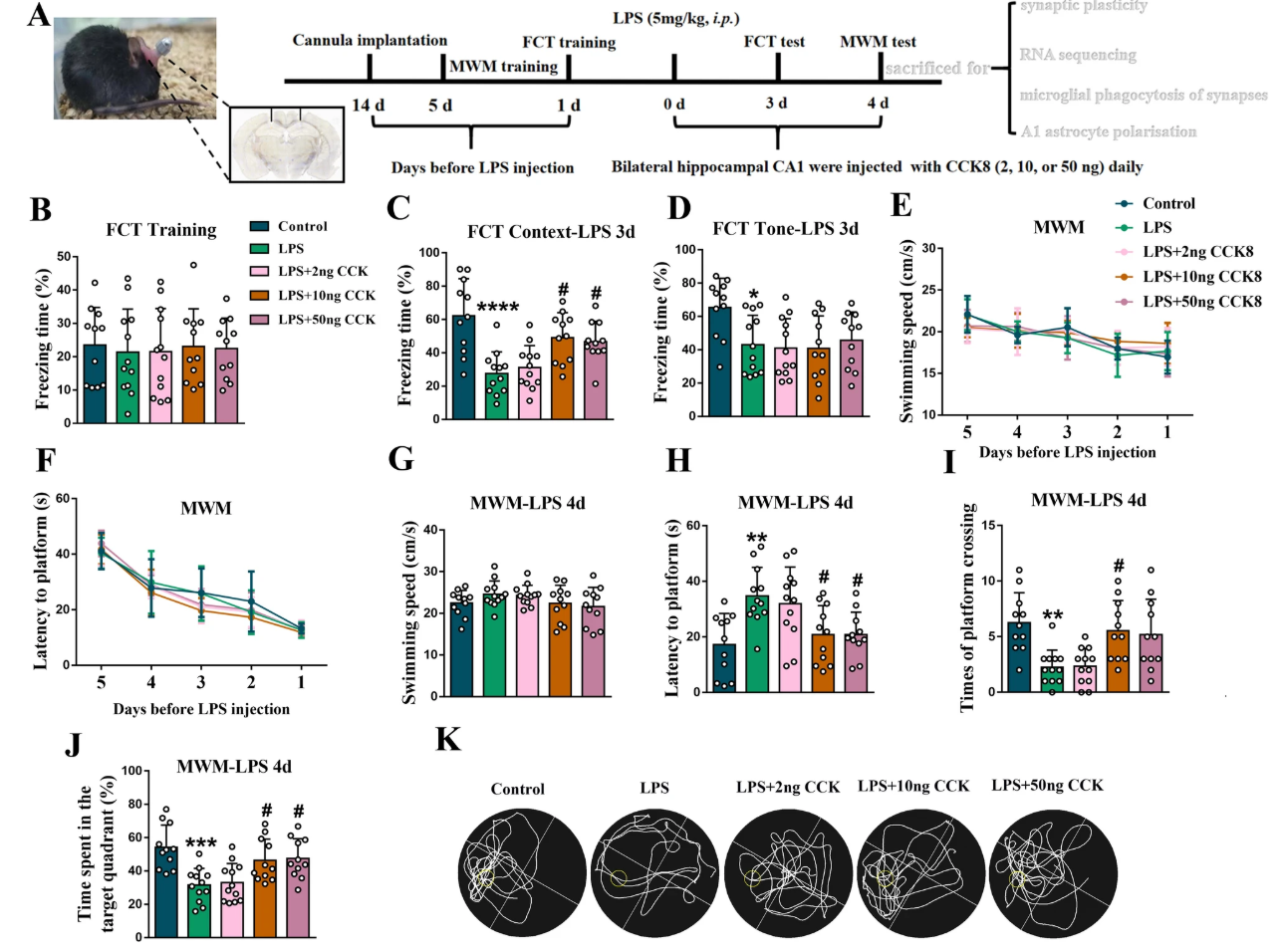
剂量筛选:10ng和50ng CCK8可显著增加SAE小鼠FCT场景测试冻结时间(改善海马依赖性记忆),但对声音测试无影响(非海马依赖性记忆未改善,与仅局部注射CA1区相关)
MWM结果:10ng CCK8可显著缩短逃避潜伏期、增加平台穿越次数和目标象限停留时间,50ng CCK8可缩短逃避潜伏期、增加目标象限停留时间,且所有剂量CCK8均不影响小鼠游泳速度(排除运动能力干扰);
后续实验选用10ng CCK8(效果最显著)。
3. CCK8可改善SAE小鼠海马CA1区的兴奋性突触可塑性
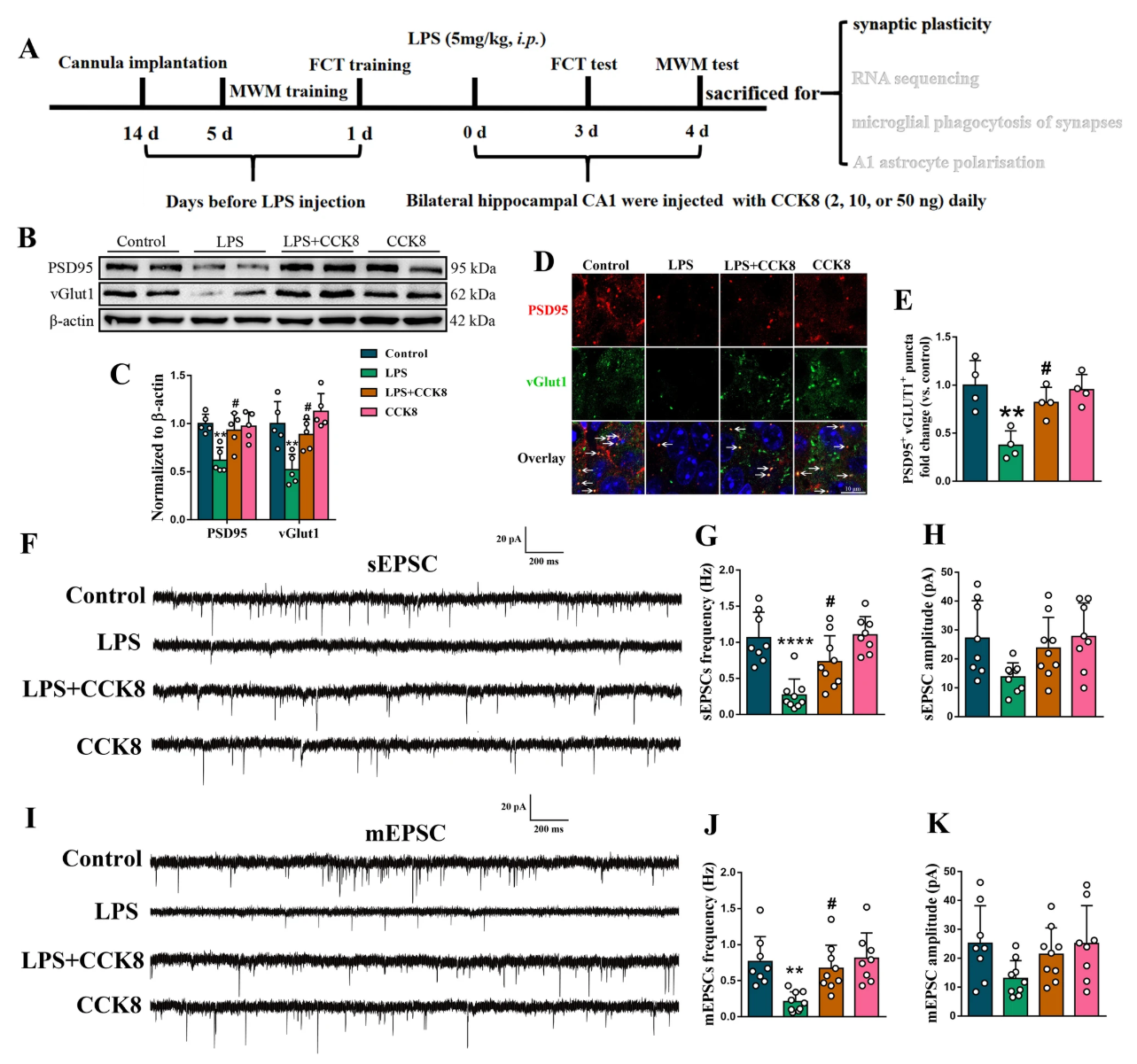
突触数量:CCK8干预后,SAE小鼠海马CA1区PSD95、vGlut1蛋白水平及兴奋性突触数量显著恢复;
突触功能:膜片钳显示,CCK8可显著增加SAE小鼠海马CA1区兴奋性神经元的sEPSCs和mEPSCs频率和振幅
4. CCK8通过抑制C1q介导的小胶质细胞突触吞噬和A1型星形胶质细胞极化发挥作用
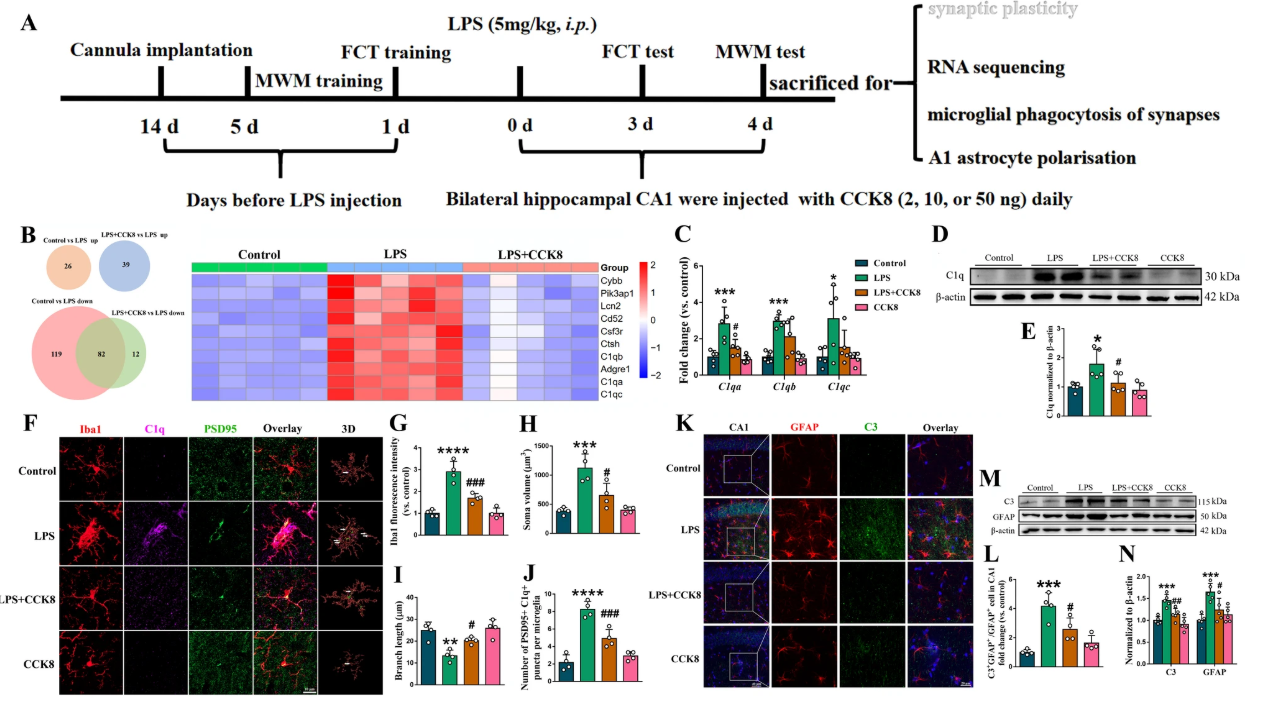
基因筛选:RNA测序显示,与LPS组相比,CCK8干预组有94个基因下调,其中82个与正常对照组下调基因重叠,C1qa/b/c(补体1q的三个亚型)为差异最显著的基因之一;
C1q表达验证:qPCR和Western blot证实,CCK8可显著降低SAE小鼠海马C1q的mRNA和蛋白水平;
小胶质细胞调控:CCK8可显著抑制SAE小鼠小胶质细胞激活(降低Iba1荧光强度、减小胞体体积、增加分支长度),同时减少小胶质细胞内C1q与突触的共定位,提示抑制C1q介导的突触吞噬;
星形胶质细胞调控:SAE小鼠海马A1型星形胶质细胞(GFAP+C3)数量显著增加,CCK8可显著降低GFAP、C3蛋白水平及二者共定位,提示抑制C1q诱导的A1型星形胶质细胞极化。
5. 激活海马CA1区CCK阳性神经元与外源性CCK8具有相似的治疗效果
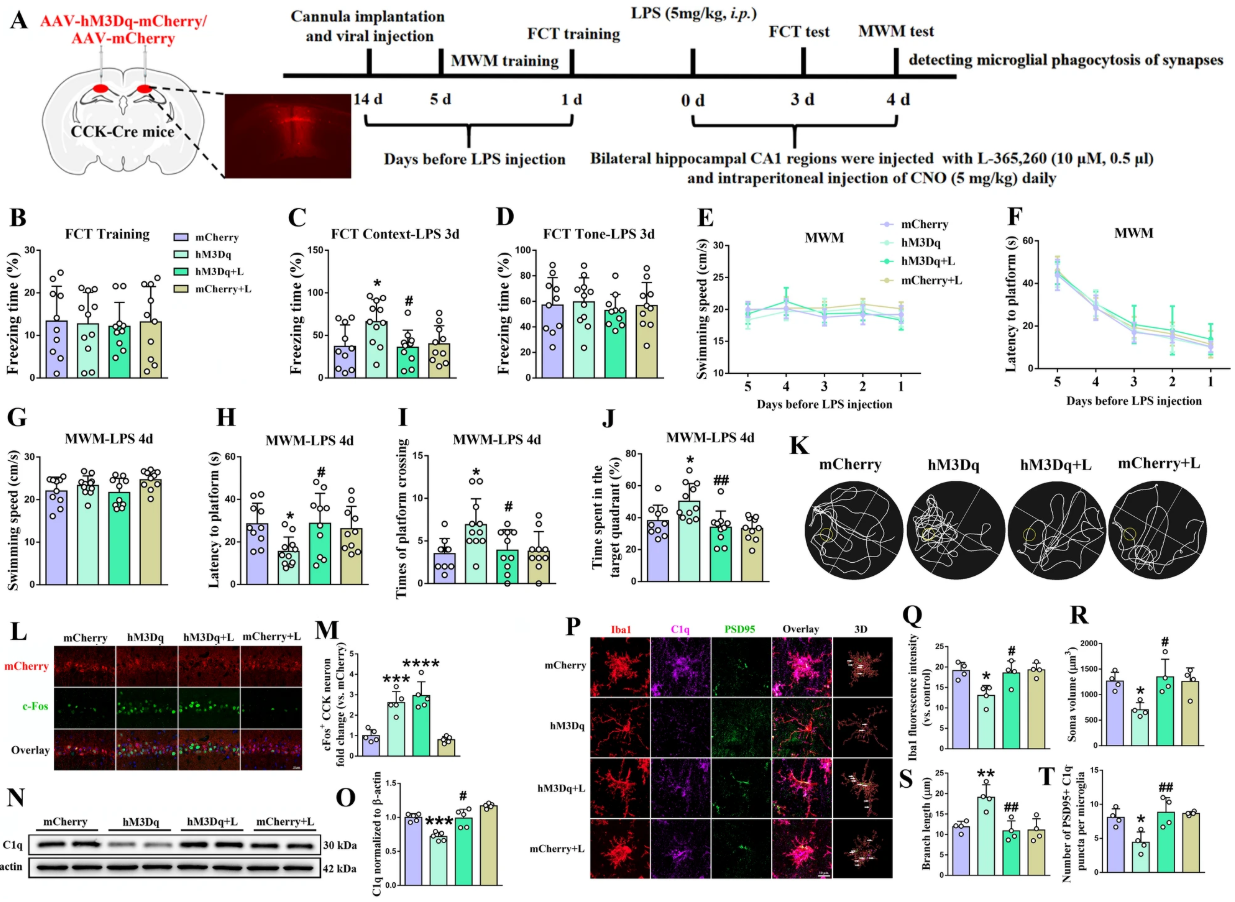
化学遗传学激活:注射激活病毒的SAE小鼠海马CA1区c-Fos阳性细胞数显著增加,且CCK2R拮抗剂L365,260不影响神经元激活(仅阻断CCK作用);
认知改善:激活CCK阳性神经元可显著增加SAE小鼠FCT场景测试冻结时间、改善MWM空间记忆,该效应可被L365,260完全阻断;
激活CCK阳性神经元可显著降低SAE小鼠海马C1q蛋白水平,抑制小胶质细胞激活和C1q介导的突触吞噬,该效应同样可被L365,260阻断。
6. 体外实验证实:CCK8通过小胶质细胞CCK2R抑制LPS诱导的C1q表达
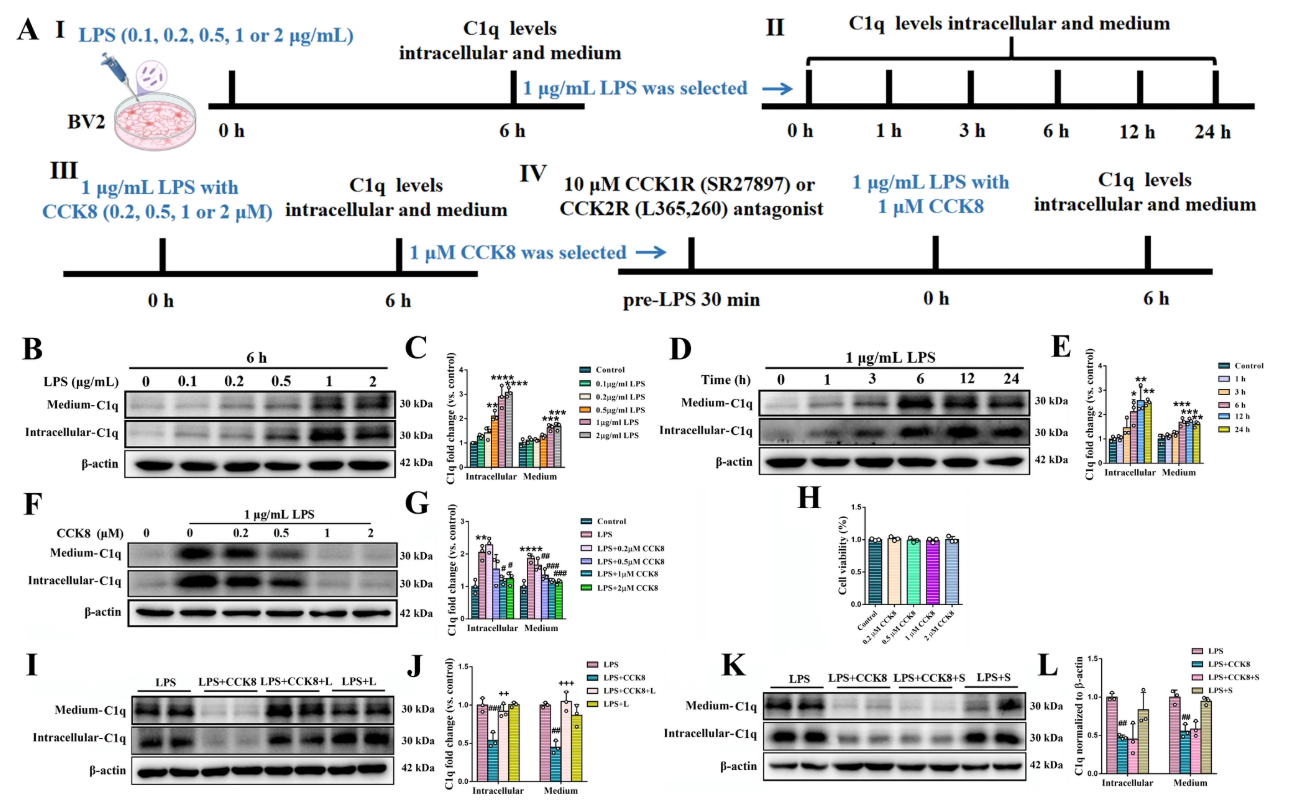
CCK8剂量效应:1μM和2μM CCK8可显著降低LPS刺激的BV2细胞内和培养基中C1q蛋白水平,且对BV2细胞活力无影响,后续选用1μM CCK8;
受体特异性:CCK2R拮抗剂L365,260可完全阻断CCK8对C1q的抑制作用,而CCK1R拮抗剂SR27897无此作用,证实CCK8通过小胶质细胞CCK2R发挥作用。
四、研究讨论
1. 核心机制总结
CCK(主要为CCK8)通过小胶质细胞CCK2R抑制C1q表达,从两个层面改善兴奋性突触可塑性:①直接抑制小胶质细胞对C1q标记的兴奋性突触的过度吞噬;②间接抑制C1q诱导的A1型星形胶质细胞极化,减少其突触毒性,最终恢复海马CA1区突触结构和功能,改善SAE小鼠的海马依赖性认知障碍(机制示意图见图7)。
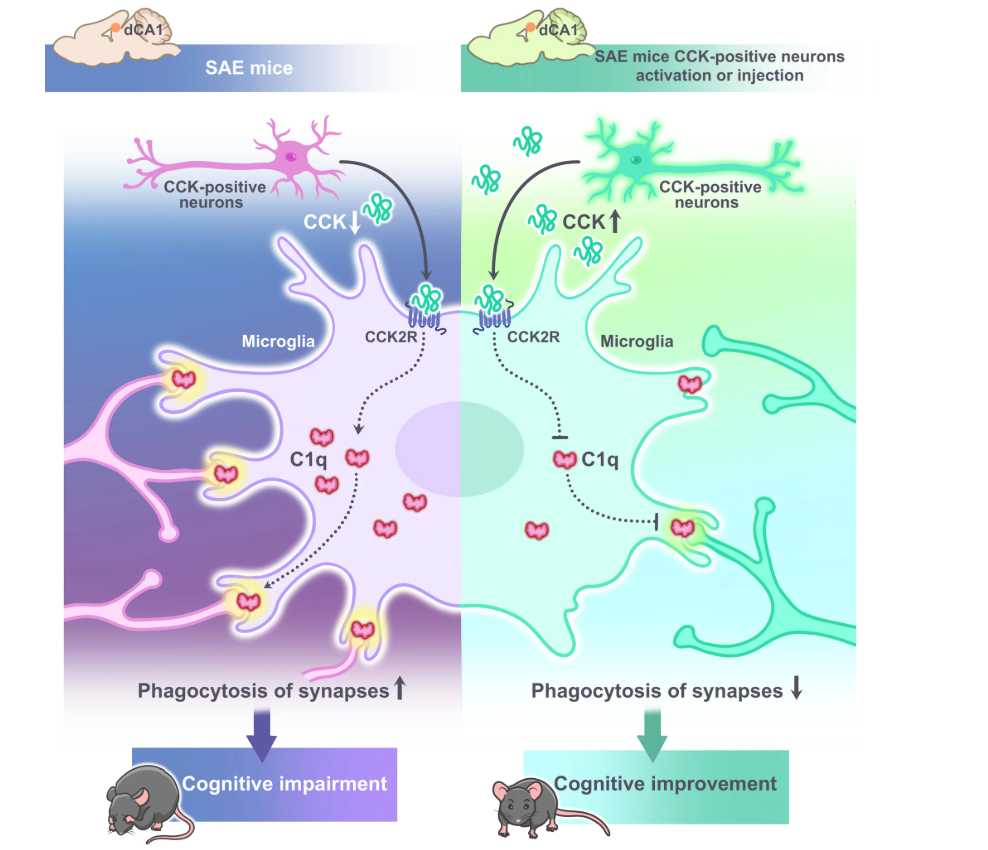
2. 研究创新性
首次将CCK与SAE关联,明确了CCK在SAE认知障碍中的保护作用;首次证实内源性CCK阳性神经元激活与外源性CCK补充具有等效治疗效果,为SAE的基因治疗提供了新方向;进一步完善了“补体-小胶质细胞-突触丢失”的SAE发病机制,明确C1q是CCK发挥作用的关键靶点;证实CCK2R是中枢介导SAE神经保护的核心受体,为高选择性CCK2R激动剂的研发提供了实验依据。
3. 研究局限性
细胞模型单一:仅使用永生化BV2小胶质细胞,未验证原代小鼠/人小胶质细胞,结果的临床转化需进一步验证;
造模为短期认知障碍:5mg/kg LPS诱导的SAE小鼠认知障碍7d可部分恢复,而临床SAE常表现为长期认知障碍,需采用盲肠结扎穿刺(CLP)或高剂量LPS造模,验证CCK对长期认知障碍的作用;
未排除其他通路:本研究聚焦C1q-小胶质细胞-突触通路,未排除CCK通过抗炎、调节神经元兴奋性等其他通路改善认知的可能;
仅研究雄性小鼠:未纳入雌性小鼠,性别差异对CCK作用的影响需进一步探索。
五、研究结论
本研究结果表明,CCK通过小胶质细胞CCK2R抑制C1q表达,从而减弱C1q介导的突触可塑性损伤,最终改善SAE模型小鼠的认知功能障碍。CCK药物和特异性激活CCK阳性神经元都是SAE的潜在治疗方法。
文献原文及链接:https://doi.org/10.1186/s12974-025-03554-9
