

Theranostics 空军医科大学第一附属医院揭示SOX18 m6A修饰通过上调PTX3促进心肌细胞焦亡和脓毒症心肌病的进展研究
脓毒症是由感染引发的全身性炎症反应,病情严重时可导致器官功能障碍和多器官衰竭。脓毒症诱导的心肌病(SIC)是指由脓毒症引起的心脏功能障碍。这是一种危及生命的急症,病情往往在短时间内迅速恶化。脓毒症引发的炎症反应会导致大量炎症介质(如趋化因子和白细胞介素)的释放,这些介质会损害心肌细胞并影响心脏功能。此外,血流动力学紊乱还会进一步扰乱其他器官的正常功能,严重损害患者的生活质量。然而,SIC 的发病机制尚未完全明了,这给治疗带来了重大挑战。
2025年2月,空军医科大学第一附属医院在Theranostic杂志发表题为“The m6A modification of SOX18 leads to increased PTX3 and cardiomyocyte pyroptosis in Sepsis-Induced Cardiomyopathy”的研究型论文,揭示了SIC心 肌 细 胞 中RBM15和YTHDF2的异常升高水平介导了SOX18的m6A修饰,导致SOX18表达下调,SOX18水平降低无法有效抑制PTX3转录,从而导致PTX3水平升高,促进炎性小体形成和细胞焦亡,进一步加剧心脏功能障碍,具有重要的临床意义。
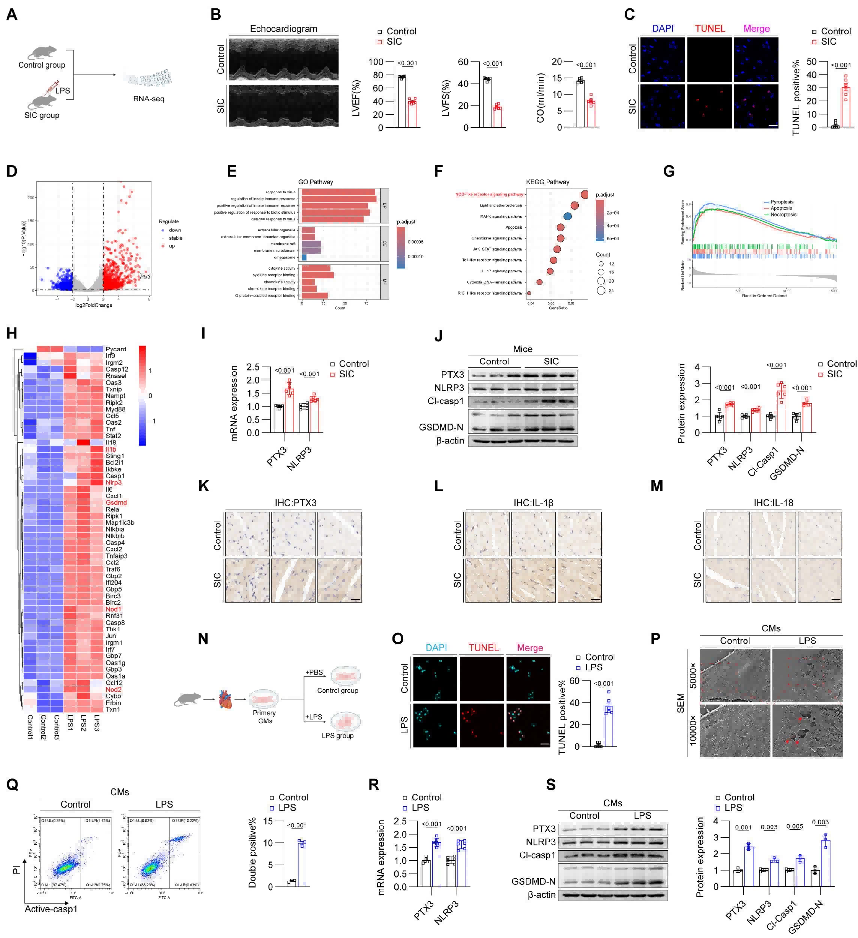
为确定SIC的潜在治疗靶点,通过小鼠SIC心肌组织测序,筛选出主要的表型变化及差异表达分子:细胞焦亡与PTX3。体内检测LPS诱导的小鼠心肌组织发现PTX3及焦亡分子表达升高,体外检测LPS处理的新生小鼠心肌细胞发现PTX3表达增加及心肌细胞死亡、焦亡加重,LPS处理下PTX3与NLRP3共定位且存在相互作用,证实PTX3参与了NLRP3炎性小体的形成。
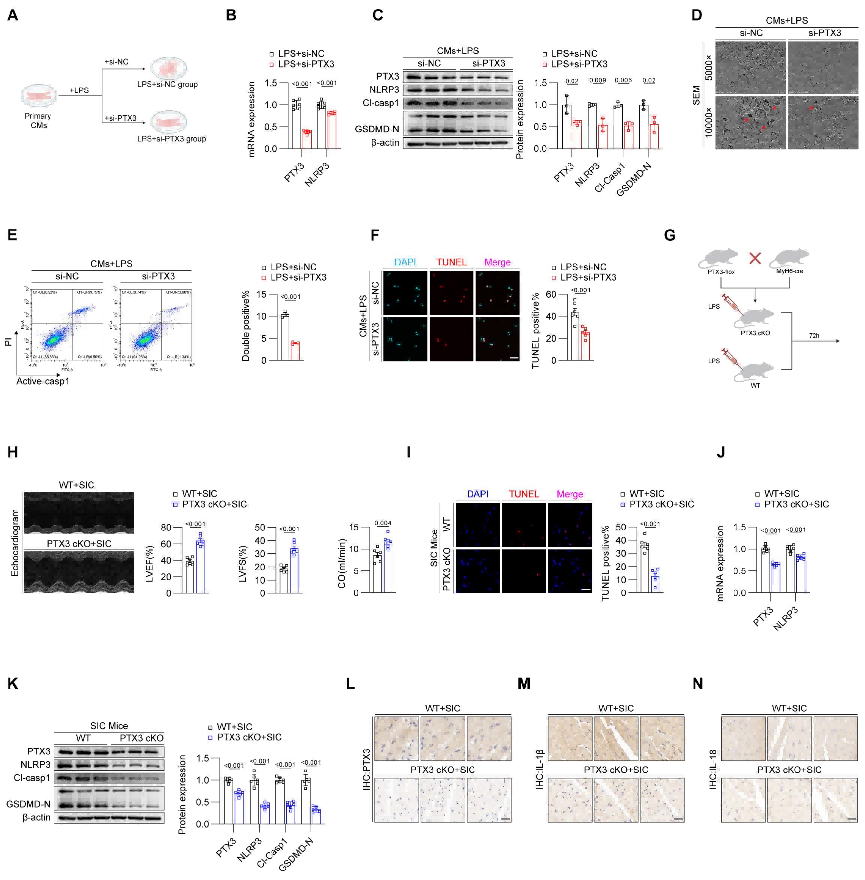
为确认 PTX3 对心肌细胞焦亡的影响,体外干扰PTX3的表达显著降低心肌细胞NLRP3、Cl-Casp1、GSDMD的表达,体外干扰PTX3的表达显著抑制心肌细胞焦亡,干扰PTX3表达能有效抑制 NLRP3 炎性小体的形成。构建心肌细胞PTX3条件性敲除小鼠,心肌细胞PTX3条件性敲除可有效缓解SIC小鼠心功能障碍,心肌细胞PTX3条件性敲除可有效抑制SIC小鼠心肌组织焦亡。
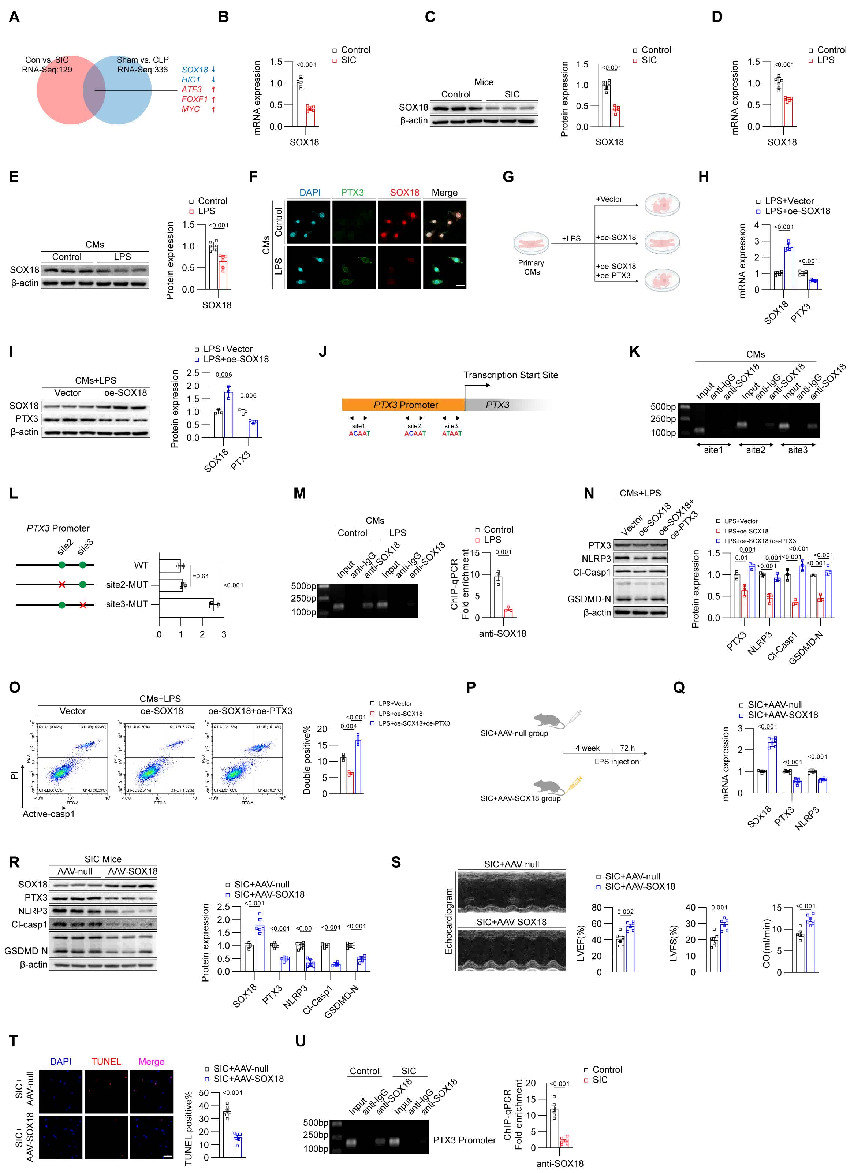
鉴于 RNA-seq 和 qPCR 分析已证实 PTX3 的增加主要发生在 mRNA 水平,着重于识别可能调控 PTX3 表达的转录因子。通过结合 CLP和 LPS 诱导的小鼠 SIC 模型的 RNA-Seq 数据以及 JASPAR 和 CISBP 等数据库,确定了五个可能的转录因子,包括 SOX18、HIC1、ATF3、FOXF1 和 MYC,它们可能调节 PTX3 的表达,其中,SOX18 的变化最为显著。ChIP及双荧光素酶报告基因检测实验证实SOX18富集于PTX3启动子区且抑制PTX3的转录活性,体外挽救实验证实SOX18介导LPS刺激下PTX3对心肌细胞焦亡的促进作用。
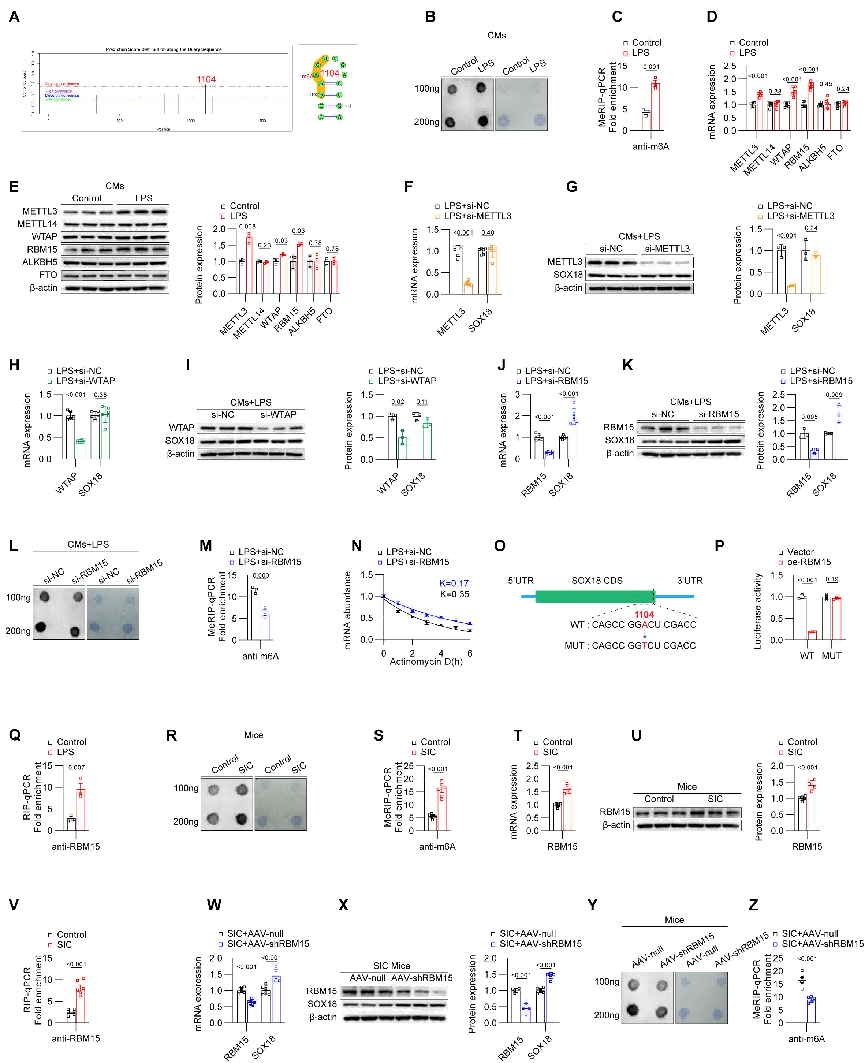
N6-甲基腺苷(m6A)修饰是一种广泛存在且具有重要功能的表观遗传标记。研究小组此前已在多种心血管疾病中发现了 m6A 修饰,例如心房颤动和糖尿病性心肌病,并证明 m6A 修饰在这些疾病的进展中起着关键作用。假设 m6A修饰可能同样会影响 SIC-SOX18-PTX3通路。为了验证这一假设,使用 SRAMP 预测工具分析了 SOX18 的 mRNA 序列,发现 SOX18 mRNA上存在潜在的 m6A 修饰位点,其中得分最高的位点位于第 1104 位的腺苷。LPS 组小鼠心肌细胞的总m6A水平显著高于对照组,敲低RBM15可显著上调SOX18的表达,而敲低其他写入酶如METTL3和WTAP对SOX18的表达无影响。敲低RBM15可显著上调SOX18的表达,而敲低其他写入酶如METTL3和WTAP对SOX18的表达无影响,SOX18 mRNA潜在的m6A修饰位点(第1104位腺苷)突变可消除RBM15对SOX18 mRNA的抑制作用,敲低SIC小鼠心肌细胞RBM15 的表达可上调SOX18,抑制整体m6A修饰水平和 SOX18 mRNA上的m6A 修饰水平。
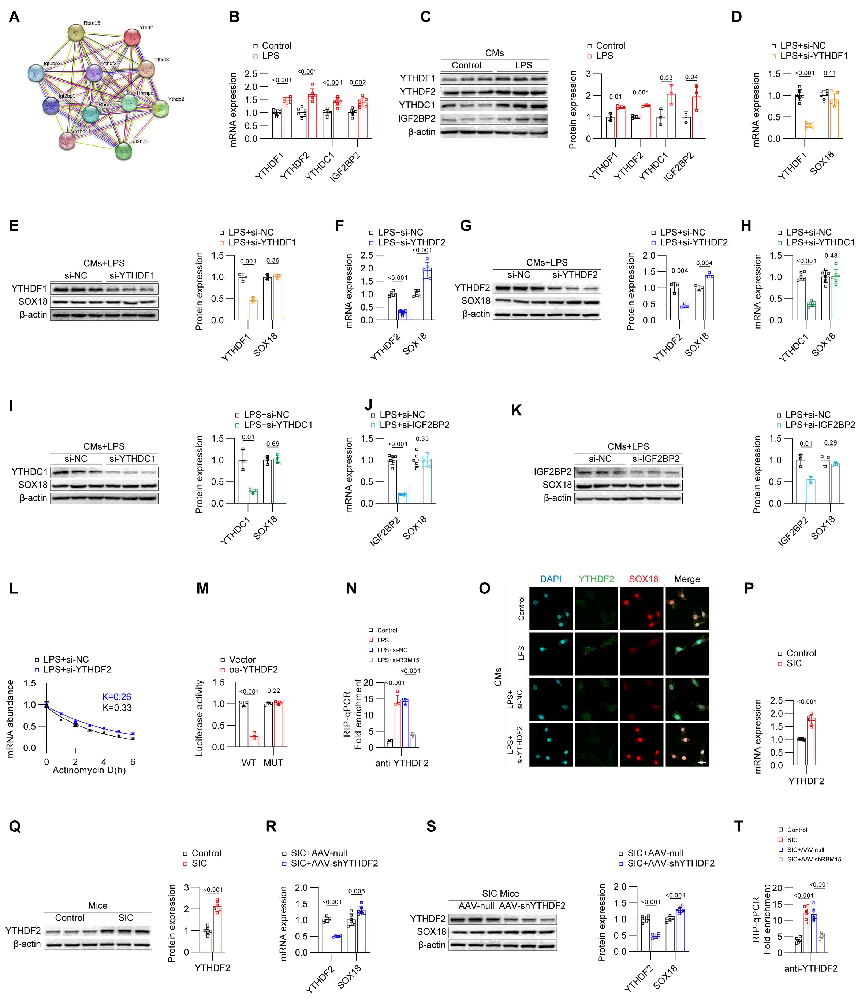
在 m6A 修饰的生物学功能中,m6A 修饰的“写入者”和“读取者”都发挥着关键作用;m6A写入者负责添加修饰,而 m6A 读取者识别这些修饰并调节相应的生物学过程。利用 STRING 数据库,确定了几个可能与 RBM15 协同作用的潜在 m6A 读取者。qPCR 和 Western blot分析显示,在脂多糖(LPS)刺激的原代小鼠心肌细 胞 中 ,YTHDF1、YTHDF2、YTHDC1 和IGF2BP2 的表达均上调。为了确定可能作用于 SOX18 的 m6A 读取者,我们用针对这些读取者的 siRNA 转染 LPS 刺激的心肌细胞。qPCR 和 Western blot 结果表明,沉默 YTHDF2 能有效提高 SOX18 的表达,而沉默其他读取者对SOX18 水平没有影响。YTHDF2干扰增强了SOX18 mRNA稳定性,SOX18 m6A修饰位点突变消除YTHDF2对SOX18 mRNA稳定性的抑制,当RBM15被沉默时,YTHDF2 与 SOX18 mRNA的结合减少,SIC小鼠心肌组织中YTHDF2表达增加,其与SOX18 mRNA结合的增加是由RBM15介导的。
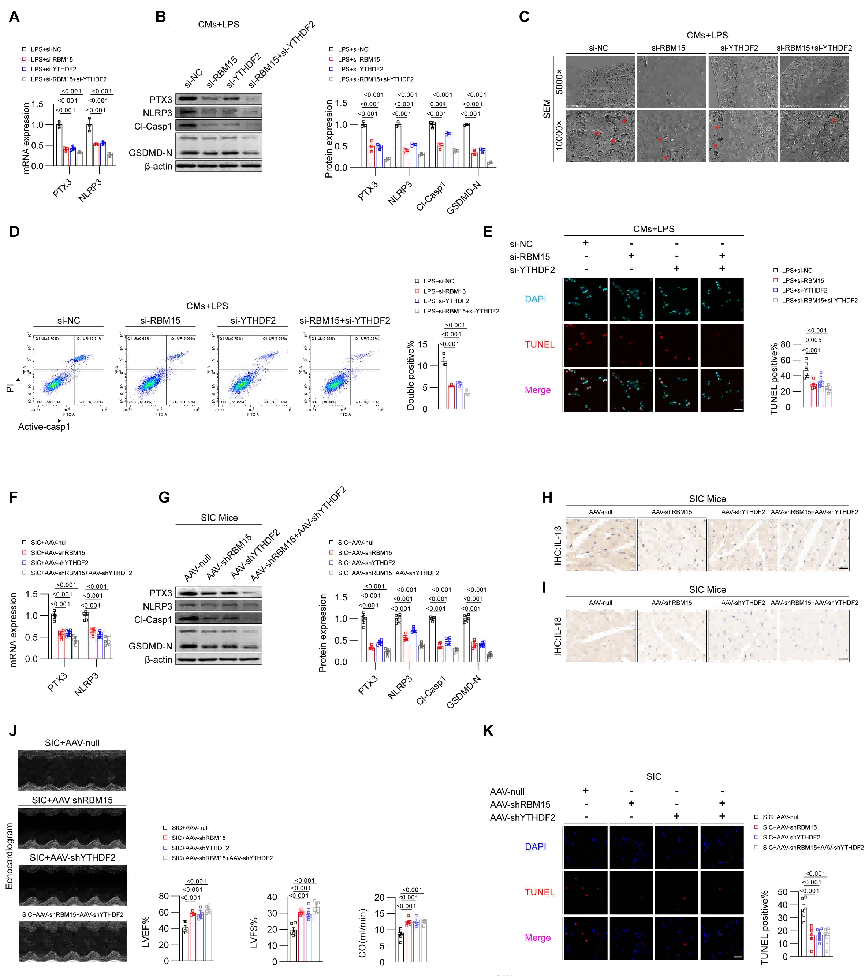
体外干扰RBM15/YTHDF2显著降低了PTX3和细胞焦亡标志物的水平、减轻与细胞焦亡相关的膜穿孔、抑制LPS诱导的心肌细胞死亡;体内干扰RBM15/YTHDF2的表达可显著抑制SIC小鼠心脏组织中细胞焦亡标志物的水平、缓解SIC诱导的心脏功能障碍、减轻心肌损伤、降低循环中细胞焦亡标志物的水平。
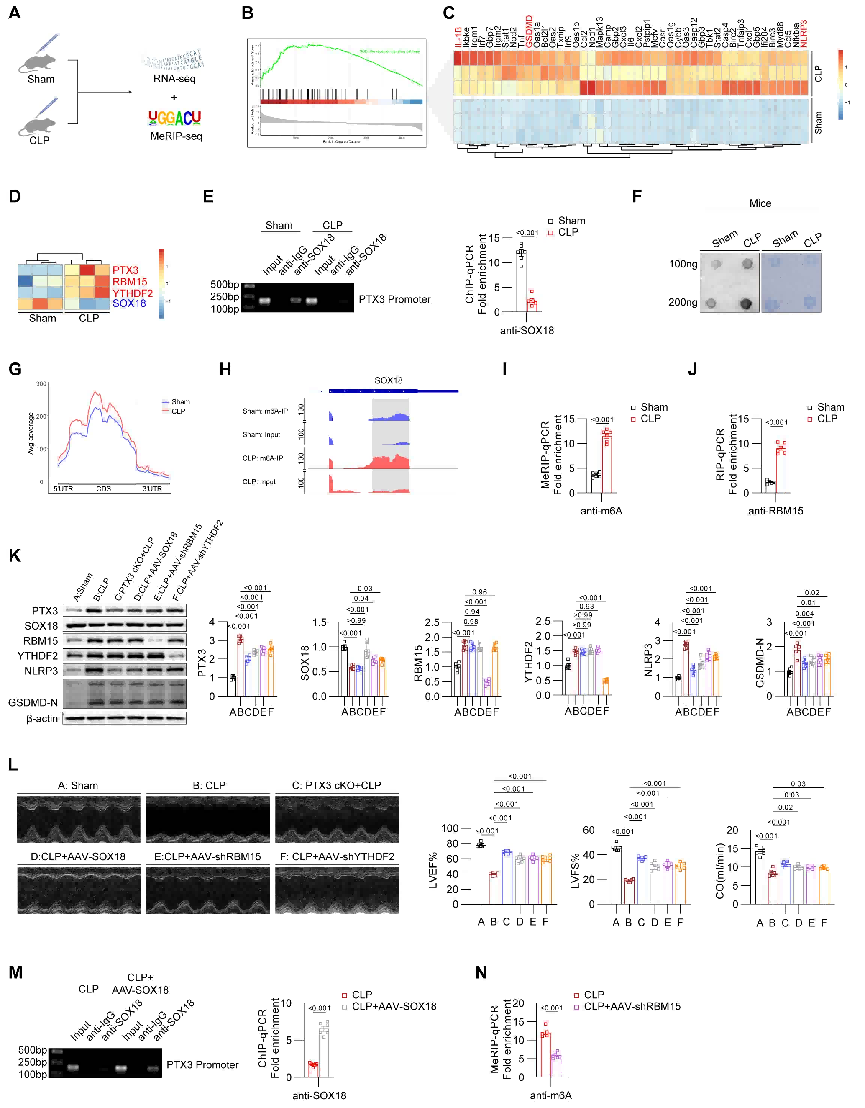
在 CLP 模型中验证了先前的研究结果:CLP 小鼠体内 RBM15 和 YTHDF2 水平升高会促 进 SOX18 的 m6A 修 饰 , 导 致 其 表 达 下 调 。SOX18 水平降低无法转录抑制 PTX3,从而引发心肌细胞焦亡和心脏功能受损。
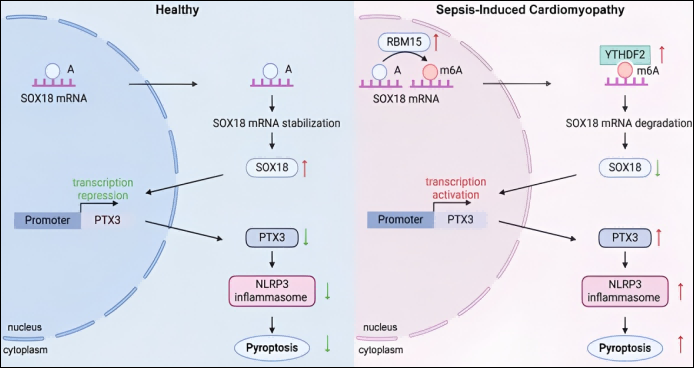
本研究结果表明,PTX3 是 NLRP3 炎性小体的关键成分。在 SIC 进展过程中,PTX3 显著上调并引发细胞焦亡反应,抑制 PTX3 能有效减轻 SIC 中的心肌细胞焦亡;其次,SIC中SOX18 的下调是导致 PTX3 表达增加的主要因素,在生理条件下,SOX18 与 PTX3 的启动子区域结合并抑制其转录,然而,在 SIC 期间,SOX18 表达减少,导致无法有效抑制 PTX3 的升高;最后,证明了 SIC 中SOX18 的下调是由RBM15/YTHDF2介导的,RBM15催化SOX18 mRNA的 m6A修饰,而YTHDF2识别这种m6A修饰,降低 SOX18 mRNA的稳定性,从而导致 SOX18 表达减少。
