


文章概述
通过一项基于挪威HUNT研究的全基因组连锁分析,探讨了遗传因素与血流感染(BSI)风险的关系。研究追踪了47个家族(共365人,其中110人患病)23年的数据,发现染色体4p14上的TLR10/1/6基因位点与BSI风险存在显著关联(LOD 3.2),其中TLR10和TLR1的两个特定SNPs(rs11466658和rs3923647)与感染风险增加相关。这些基因编码的Toll样受体在先天免疫中起关键作用,其变异可能影响对血流感染的易感性。
一、研究背景
血流感染(BSI)死亡率高,早期识别容易发生BSI的受试者是很重要的。环境风险因素(肥胖、吸烟、缺乏体力活动和铁缺乏等)与BSI相关 vs. 遗传易感性(具有挑战性)。既往全基因组关联研究(GWAS)对感染性疾病的遗传解析存在局限,主因罕见变异难以检测及表型异质性。连锁分析利用来自家庭的遗传数据来评估DNA区域是否与所关注的性状相关联,而研究多为无遗传相关人群。该研究利用可用的基因型数据重建了47个多代家系,并在1995—2017年随访的人群队列中进行了全基因组连锁分析,以及随后的基于家系的关联检测,为感染遗传学提供了新视角。
二、材料与方法
1. 研究人群
基于挪威北特伦德拉格省健康研究(HUNT)的队列研究基础(n=71860);随访23年(1995年至2017年)的血培养数据。
2. 遗传数据和质量控制
基因分型、样本和变异质量控制与填补
(1)使用三种不同的人类基因分型芯片进行基因分型,样本和单核苷酸多态性(SNP)的检测率设定为高于99%,无关样本间符合哈迪-温伯格平衡(群体遗传平衡定律)的阈值为P值小于0.0001。
选取存在两名及以上二级亲属或更近亲缘关系且感染过BSI的家系样本)
(3)目标家系的样本和SNP质控(SNPs=146044)
排除未受影响的远亲、孟德尔遗传错误标记、次要等位基因频率(MAF)低于0.1%的SNPs、PLINK软件二次筛选)。
三、分析
本研究仅对常染色体进行分析。使用Merlin软件对146,044个直接基因分型的SNP位点进行了多点非参数连锁分析。我们通过计算对数率(LOD)得分来评估基因位点与疾病的连锁关系。为了减少连锁不平衡导致的膨胀关联,我们仅评估符合以下标准的SNP:如果R2大于0.1且物理距离小于10 kb,我们将snp聚集在全基因组范围内,共计25,724个集群。为了纳入来自未患病家族成员的信息,并试图缩小关注区域,对显示峰区域和BSI之间最强关联的家族内填补变异体集进行了基于家族的关联检验(选择了峰区域内家族内LOD阳性(LOD>0.01)的家族),进而追踪了最显著的连锁峰。敏感性分析:探讨在家庭内是否存在的BSI累积。
四、结果
(一)一般特征
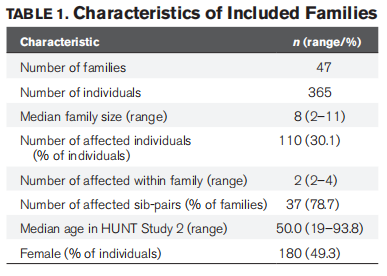
对69,423名通过质控的基因型受试者进行了分析,确定47个家庭中至少有两名二级亲属或更近亲属感染了BSI,这47个家庭共包含365名基因型个体,其中110人确诊血流感染。
(二)基因组连锁分析
|
|
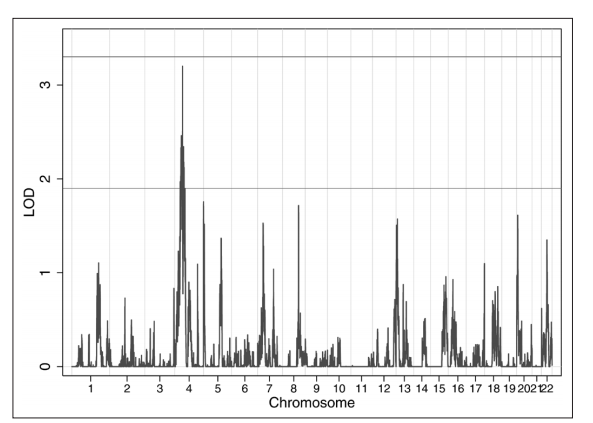
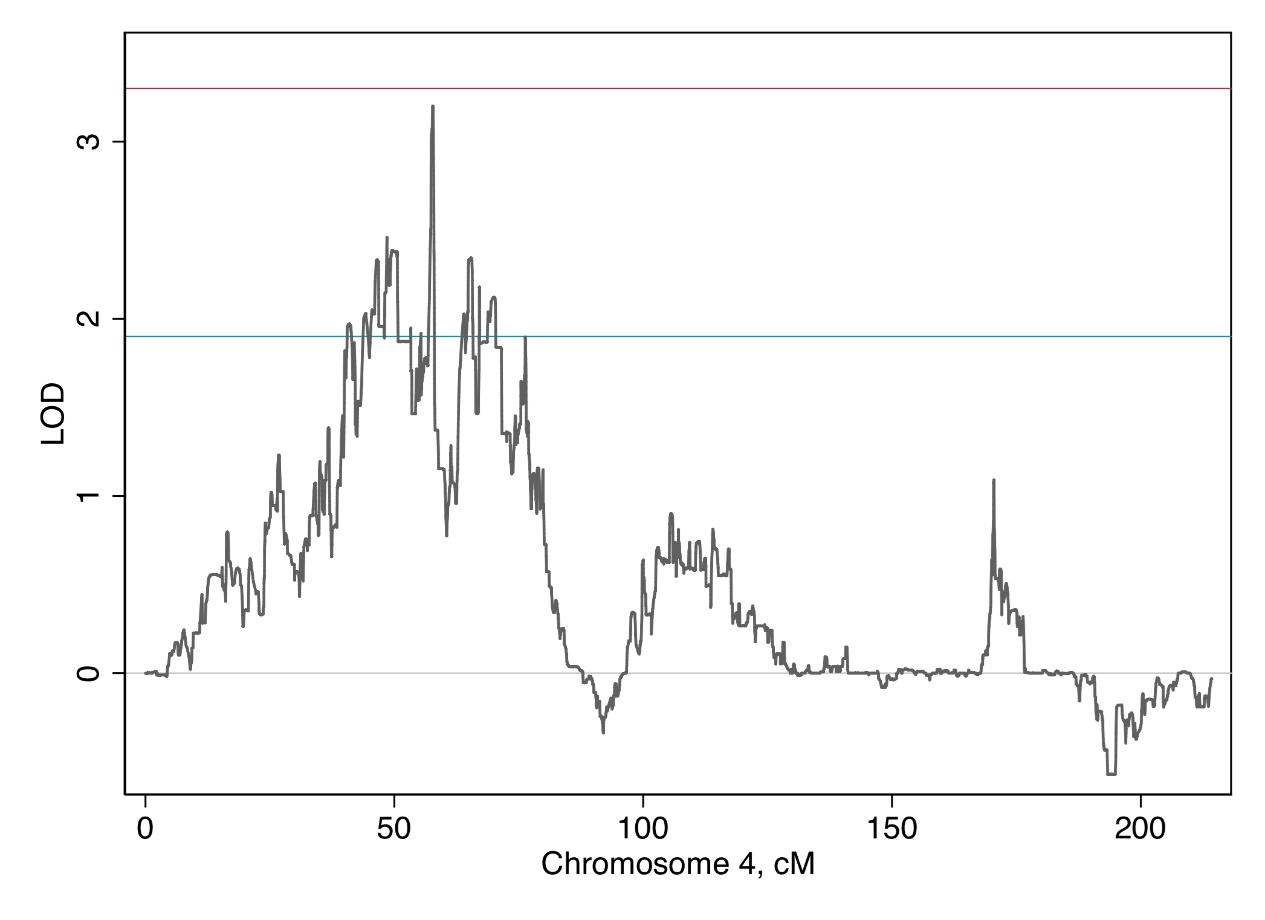
图1全基因组连锁分析血流感染(BSI)风险 图2 4号染色体与血液感染风险的连锁分析
在全基因组连锁分析中,没有一个位点达到全基因组显著性(LOD > 3.3),但存在三个提示性位点(LOD >1.9),均位于4号染色体上,分别位于46.6cM(LOD2.3)、57.7cM(LOD 3.2)和70.0cM(LOD 2.1)。蓝线和红线分别表示提示性和全基因组显著的对数概率(LOD)评分。
(三)敏感性分析
当峰值区域的LOD值下降1个单位时,可确定57.1至58.0厘摩范围内的区域。该区域内共有七个基因(从染色体起始位置开始计数):FLJ13197、KLF3、TLR10、TLR1、TLR6、FAM114A1和MIR574。
在峰值区域最显著的0.1 LOD下降点(57.6-57.8厘摩),可识别出三个基因:TLR10、TLR1和TLR6。
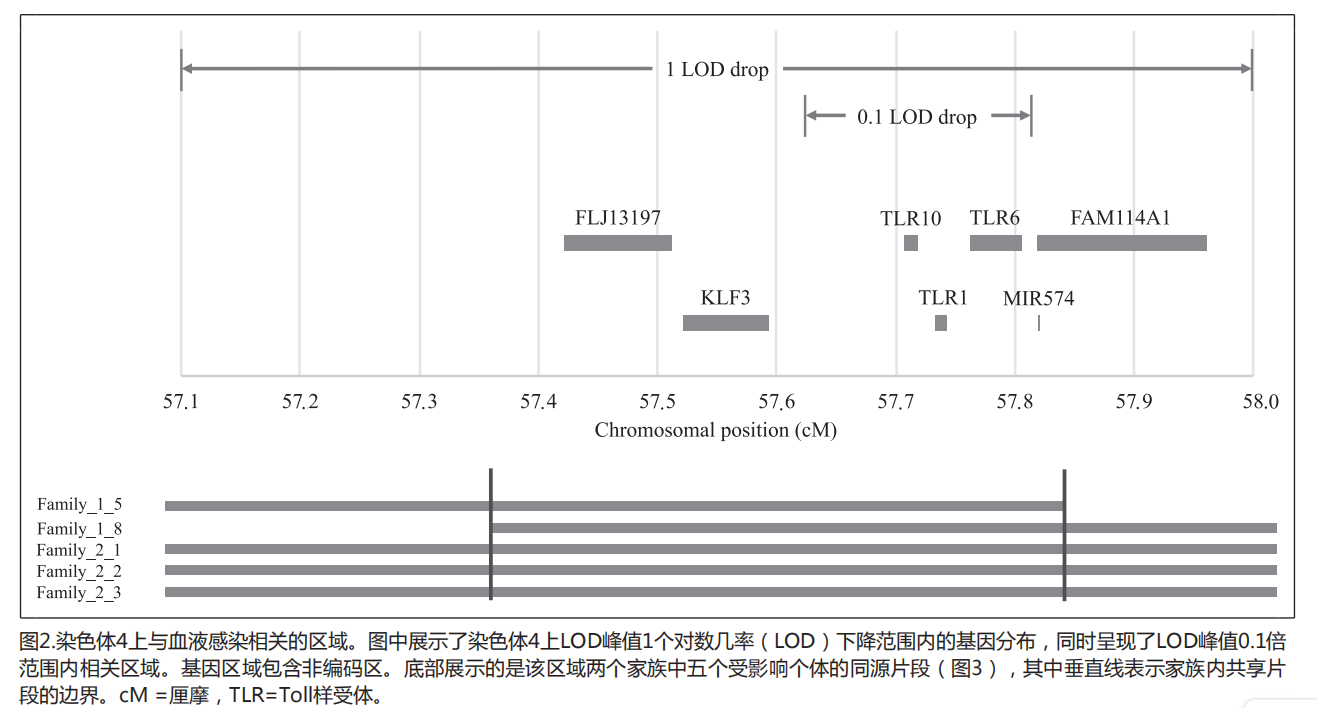
图3显示染色体4上的区域与血流感染相关。
(四)家系关联分析
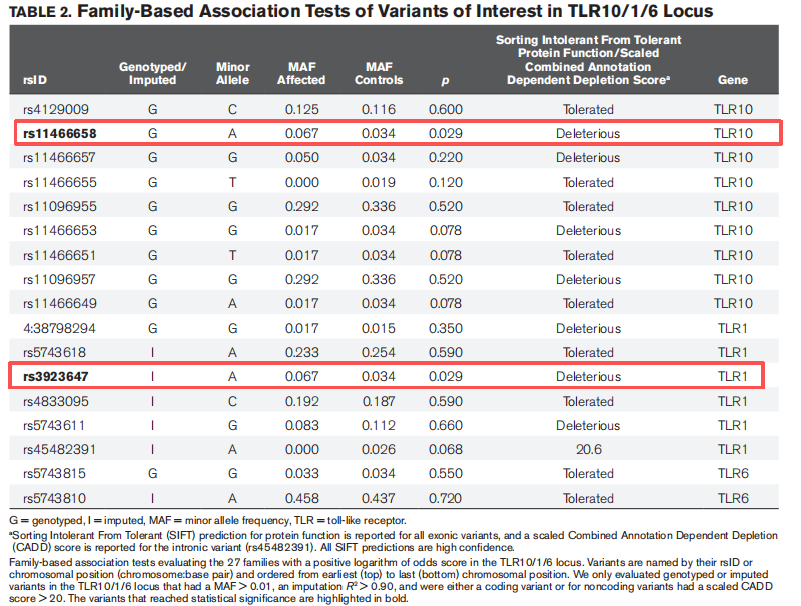
在47个家庭中,选取LOD评分在峰区呈阳性的27个家庭(60例和134例对照)进行关联分析。
两个SNP具有统计学显著性:TLR10基因中的rs11466658和TLR1基因中的rs3923647。这两种突变均被预测会对蛋白质功能产生有害影响,且病例组的等位基因频率约为对照组的两倍。
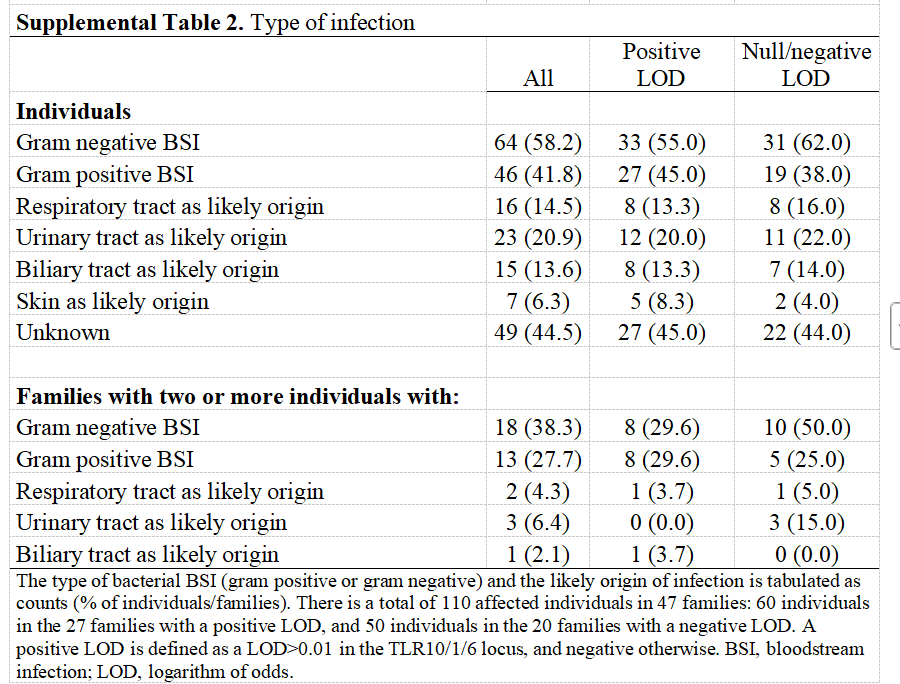
(五)评估在峰值位点检测结果为阳性/阴性(LOD)的家族感染类型的分布情况
与阴性LOD的家族相比,阳性LOD家族在BSI感染类型及感染源方面均未呈现明显模式。
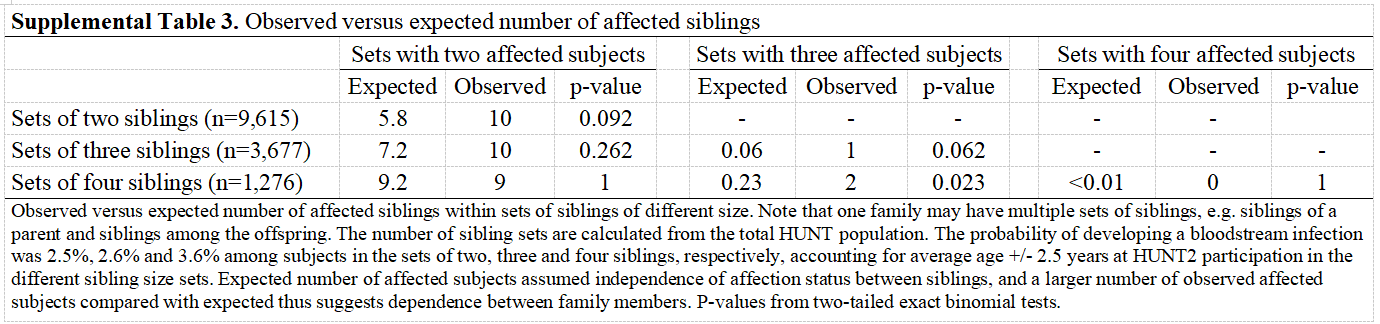
(六)评估兄弟姐妹集内BSI的积累情况
BSI事件在同胞组内聚集的趋势明显。
值得注意的是,在四个兄弟姐妹集中,有三个受影响受试者的集合的预期数量为0.23,观察到的集合数量为2 (P = 0.023)。
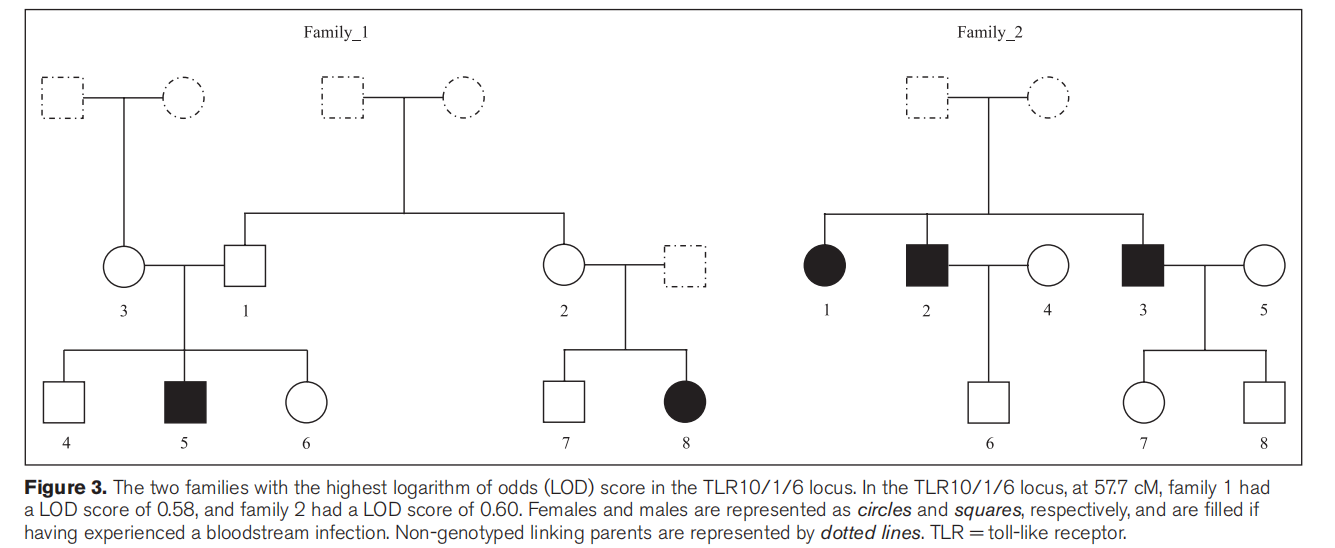
(七)TLR10/1/6基因座中具有最高对数优势比评分的两个家族
在TLR10/1/6位点上对数率(LOD)评分最高的两个家系。
在TLR10/1/6位点,在57.7 cM处,家系1的LOD值为0.58,家系2的LOD值为0.60。女性和男性分别表示为圆形和正方形,如果经历过血流感染则填充。非基因型连接亲本用虚线表示。
五、结论
在全基因组连锁分析及后续的家庭为基础的关联检测中发现,4p14染色体的变异,可能与TLR10/1/6基因座的突变相关,且与在以挪威人为基础的群体样本中发展BSI的风险增加有关。
六、创新性和不足
(一)创新性
1. 首个在前瞻性队列中评估发生BSI风险的全基因组分析。
2. 首个基于家系的BSI基因组连锁分析。
(二)不足
1. 统计效能不足(仅47家系,LOD未达全基因组显著)
2. BSI异质性(病原体/感染来源未分层分析)
3. 仅欧洲人群,需其他族群验证
文献原文:
